 基本信息
基本信息
 工作经历
工作经历
 研究方向
研究方向
 获奖情况
获奖情况
 代表论文
代表论文
倪挺团队开发新型单细胞聚类和批次效应矫正网络分析方法
单细胞RNA-seq技术可揭示细胞间异质性,发现新的功能亚群,解析细胞谱系分化,发现疾病新标志物等,具有重要价值并成为遗传学研究的重要前沿。2021年2月22日,分子生物学领域国际知名期刊《核酸研究》(Nucleic Acids Research)在线发表了人类遗传学与人类学系倪挺课题组题为“Independent component analysis based gene co-expression network inference (ICAnet) to decipher functional modules for better single-cell clustering and batch integration”的方法学研究论文。该研究开发了基于独立成分分析策略的基因共表达网络推断(ICAnet)方法,将单细胞表达数据分解为一系列独立的基因表达成分和推断的共表达模块,从而改善了单细胞分群性能,帮助发现具有生物学意义的稀有细胞亚群,并且可校正多种来源的批次效应,为单细胞RNA-seq的数据分析提供了一种新选择。
为了充分揭示单细胞表达数据的异质性,前人已经开发了多种单细胞聚类算法,例如Seurat、SC3、pcaReduce和SINCERA等。这些算法之间的共享策略是将单细胞表达数据投影到较低维度的空间中,并基于细胞与细胞在低维空间中的距离来进行细胞聚类。然而,这些方法通常基于单个基因来测量细胞相似性,而忽略了信号通路和/或基因表达调控网络(一组相互作用的基因)在细胞状态决定中所起的重要作用。为了克服这一缺点,一些计算方法整合了基因调控信息,以辅助单细胞RNA-seq数据的细胞聚类和功能解释。例如,SCENIC整合了从随机森林模型和推定转录因子(TF)结合位点推断出的基因共表达信息,以促进细胞类型预测和生物学解释。而SCORE使用已知的蛋白质-蛋白质相互作用(PPI)来修正基因共表达网络,从而产生更具有生物学意义的网络。尽管这些基于网络的细胞聚类方法对单细胞转录组的研究做出了重大贡献,但仍缺乏对真实组织/器官中细胞异质性及复杂性质的全面理解。当前使用的大多数方法通过计算基因与基因的相关性来反映所有细胞之间的共表达来推断基因表达模块。然而,基因表达调控是高度背景相关的。因此,基于常规相关性的方法可能会忽略仅存在于一部分细胞中的局部共表达效应,从而导致无法利用单细胞表达数据发现某些重要但稀有的细胞亚群。
为了解决这一问题,本研究团队开发了一种称为“ICAnet”的计算方法(基于独立成分分析的网络推断),以破译功能相关的基因共表达模块,从而提高了单细胞RNA-seq数据分析中细胞聚类的性能,并有效降低了不同文库类型、测序深度及单细胞数目等来源的批次效应对生物学解读的影响。ICAnet使用独立成分分析来推断不同批次之间的共享和特定表达模式,并且还结合了蛋白互作网络或转录因子-靶基因调控网络来发现跨不同数据集的“激活的”子网络(或模块),从而获得与其他算法相比综合性能更好的细胞聚类结果。这一方法的准确性、可扩展性、鲁棒性和可重复性在多个细胞系、小鼠造血细胞发育、小鼠脑皮层、人类大脑细胞、人类胰腺等10多个公共高质量单细胞数据集中得到了验证。此外还在与南京医科大学郑科教授合作产生的小鼠全睾丸单细胞RNA-seq中进行了应用。值得一提的是,运用ICAnet分析方法不仅能够找到以前的算法尚未发现但后续可以被实验验证的新型稀有细胞类型,还在急性髓系白血病单细胞RNA-seq数据中发现基因共表达模块具有作为病人预后新标记的潜能。ICAnet作为一种单细胞RNA-seq分析新方法,可在https://github.com/WWXkenmo/ICAnet/ 免费下载并使用。
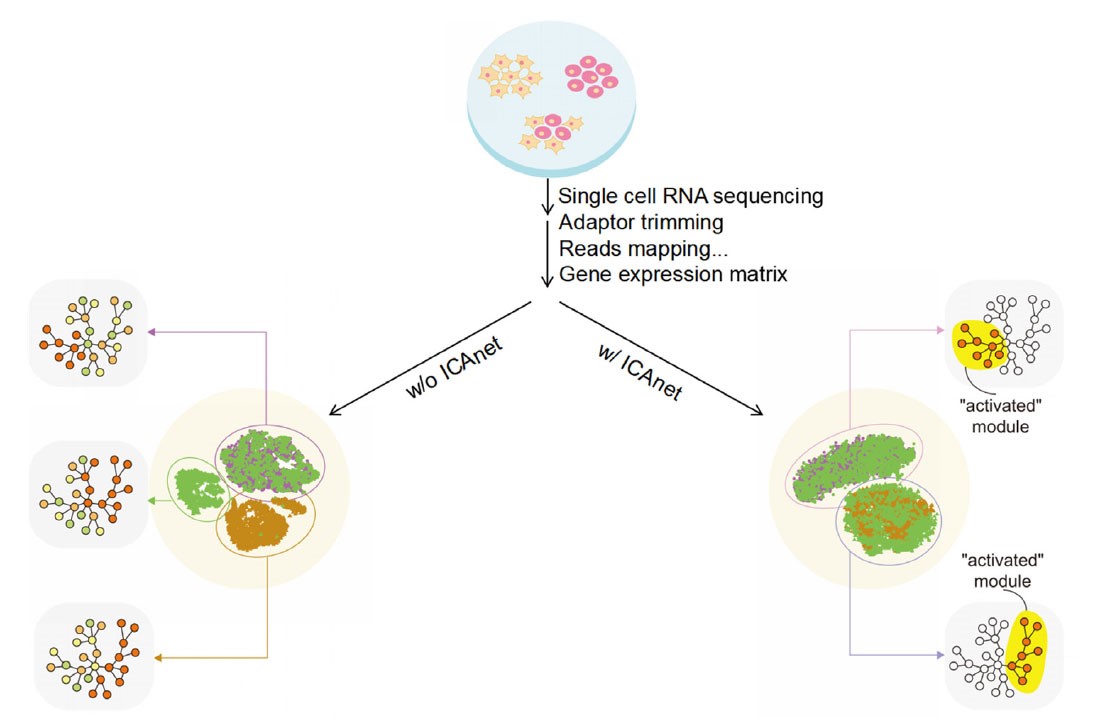
ICAnet分析方法的效果示意图
复旦大学生命科学学院倪挺课题组硕士生汪伟旭为本文的第一作者,倪挺教授、复旦大学生命科学学院魏刚博士和南京医科大学郑科教授为论文的共同通讯作者。该研究获得国家重点研发计划课题、国家自然科学基金项目及上海市首批市级科技重大专项“国际人类表型组计划(一期)”的支持。
论文连接:https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab089/6146629